змЕЈЙЬДМВтЖЈЪдМСзЂВсММЪѕЩѓВщжИЕМддђ
БОжИЕМддђжМдкжИЕМзЂВсЩъЧыШЫЖдзмЕЈЙЬДМЃЈTotal cholesterolЃЌTCЃЉВтЖЈЪдМСзЂВсЩъБЈзЪСЯЕФзМБИМАзЋаДЃЌЭЌЪБвВЮЊММЪѕЩѓЦРВПУХЩѓЦРзЂВсЩъБЈзЪСЯЬсЙЉВЮПМЁЃ
БОжИЕМддђЪЧЖдзмЕЈЙЬДМВтЖЈЪдМСЕФвЛАувЊЧѓЃЌЩъЧыШЫгІвРОнВњЦЗЕФОпЬхЬиадШЗЖЈЦфжаФкШнЪЧЗёЪЪгУЃЌШєВЛЪЪгУЃЌашОпЬхВћЪіРэгЩМАЯргІЕФПЦбЇвРОнЃЌВЂвРОнВњЦЗЕФОпЬхЬиадЖдзЂВсЩъБЈзЪСЯЕФФкШнНјааГфЪЕКЭЯИЛЏЁЃ
БОжИЕМддђЪЧЙЉЩъЧыШЫКЭЩѓВщШЫдБЪЙгУЕФжИЕМЮФМўЃЌВЛЩцМАзЂВсЩѓХњЕШааеўЪТЯюЃЌврВЛзїЮЊЗЈЙцЧПжЦжДааЃЌШчгаФмЙЛТњзуЗЈЙцвЊЧѓЕФЦфЫћЗНЗЈЃЌвВПЩвдВЩгУЃЌЕЋгІЬсЙЉЯъЯИЕФбаОПзЪСЯКЭбщжЄзЪСЯЁЃгІдкзёбЯрЙиЗЈЙцЕФЧАЬсЯТЪЙгУБОжИЕМддђЁЃ
БОжИЕМддђЪЧдкЯжааЗЈЙцЁЂБъзМЬхЯЕМАЕБЧАШЯжЊЫЎЦНЯТжЦЖЈЕФЃЌЫцзХЗЈЙцЁЂБъзМЬхЯЕЕФВЛЖЯЭъЩЦКЭПЦбЇММЪѕЕФВЛЖЯЗЂеЙЃЌБОжИЕМддђЯрЙиФкШнвВНЋЪЪЪБНјааЕїећЁЃ
вЛЁЂЪЪгУЗЖЮЇ
БОжИЕМддђЫљжИЕФзмЕЈЙЬДМВтЖЈЪдМСЪЧжИРћгУCHOD-PAPЗЈЖдШЫбЊЧхЁЂбЊНЌЕШбљБОжазмЕЈЙЬДМКЌСПНјааЬхЭтЖЈСПВтЖЈЕФЪдМСЁЃ
ФПЧАЃЌзмЕЈЙЬДМВтЖЈвбгаЭъећЕФВЮПМЯЕЭГЃКЦфОіЖЈадЗНЗЈЮЊЭЌЮЛЫиЯЁЪЭжЪЦзЗЈЃЌВЮПМЗНЗЈЮЊе§МКЭщГщЬсL-BЗДгІЯдЩЋЗЈЃЈALBKЗЈЃЉЛђЩЋЦзЗЈЃЌГЃЙцВтЖЈЗНЗЈЮЊУИЗЈЁЃУИЗЈжївЊАќРЈЕЈЙЬДМбѕЛЏУИЗЈКЭЕЈЙЬДМЭбЧтУИЗЈЃЌЦфжаЕЈЙЬДМбѕЛЏУИ-PAPЗЈЪЧФПЧАгІгУзюЮЊЙуЗКЕФГЃЙцВтЖЈЗНЗЈЁЃвРОнYY/T 1227-2014ЁЖСйДВЛЏбЇЬхЭтеяЖЯЪдМСЃЈКаЃЉУќУћЁЗЕФвЊЧѓЃЌНЋЕЈЙЬДМбѕЛЏУИ-PAPЗЈЙцЗЖБэЪіЮЊCHOD-PAPЗЈЁЃБОжИЕМддђЪЪгУгкВЩгУCHOD-PAPЗЈВтЖЈЕФЪдМСЃЌВЛЪЪгУгкЭбЧтУИЗЈКЭИЩЛЏбЇЗЈВтЖЈЕФЪдМСЃЌЕЋЪЪгУДІПЩВЮеежДааЁЃ
вРОнЁЖЬхЭтеяЖЯЪдМСзЂВсЙмРэАьЗЈЁЗЃЈЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжСюЕк5КХЃЉЁЂЁЖЪГЦЗвЉЦЗМрЙмзмОжЙигкгЁЗЂЬхЭтеяЖЯЪдМСЗжРрзгФПТМЕФЭЈжЊЁЗЃЈЪГвЉМраЕЙмЁВ2013ЁГ242КХЃЉЃЌзмЕЈЙЬДМВтЖЈЪдМСЪєгкѕЅРрМьВтЪдМСЃЌЙмРэРрБ№ЮЊЂђРрЃЌЗжРрБрТыЮЊ6840ЁЃБОжИЕМддђЪЪгУгкНјааВњЦЗзЂВсКЭЯрЙиаэПЩЪТЯюБфИќЕФВњЦЗЁЃ
ЖўЁЂзЂВсЩъБЈзЪСЯвЊЧѓ
ЃЈвЛЃЉзлЪізЪСЯ
злЪізЪСЯжївЊАќРЈВњЦЗдЄЦкгУЭОЁЂВњЦЗУшЪіЁЂгаЙиЩњЮяАВШЋадЫЕУїЁЂВњЦЗжївЊбаОПНсЙћЕФзмНсКЭЦРМлвдМАЭЌРрВњЦЗЩЯЪаЧщПіЕШФкШнЃЌгІЗћКЯЁЖЬхЭтеяЖЯЪдМСзЂВсЙмРэАьЗЈЁЗКЭЁЖЙигкЙЋВМЬхЭтеяЖЯЪдМСзЂВсЩъБЈзЪСЯвЊЧѓКЭХњзМжЄУїЮФМўИёЪНЕФЙЋИцЁЗЃЈЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙЋИц2014ФъЕк44КХЃЉЕФЯрЙивЊЧѓЁЃ
1.ВњЦЗдЄЦкгУЭО
змЕЈЙЬДМЪЧжИбЊвКжаИїжЌЕААзЫљКЌЕЈЙЬДМжЎзмКЭЃЌЗжЮЊѕЅЛЏаЭЕЈЙЬДМЃЈгжГЦЕЈЙЬДМѕЅЃЌCEЃЉКЭгЮРыаЭЕЈЙЬДМЃЈFCЃЉЃЌЦфжаCEеМ60%~70%ЃЌFCеМ30%~40%ЃЌНЁПЕИіЬхжЎМфСНжжРраЭЕФБШР§БЃГжЮШЖЈЁЃЕЈЙЬДМЪЧКЯГЩЩіЩЯЯйЦЄжЪМЄЫиЁЂадМЄЫиЁЂЕЈжЫсвдМАЮЌЩњЫиDЕШЩњРэЛюадЮяжЪЕФживЊдСЯЃЌвВЪЧЙЙГЩЯИАћФЄЕФжївЊГЩЗжЃЌЦфХЈЖШПЩзїЮЊжЌДњаЛЕФжИБъЁЃ
ЖЈСПВтЖЈШЫЬхбљБОжазмЕЈЙЬДМКЌСПЃЌСйДВжївЊгУгкаФбЊЙмМВВЁЕФЮЃЯеЗжЮіЃЌИпTCбЊжЂЪЧЙкаФВЁЕФжївЊЮЃЯевђЫижЎвЛЁЃВЁРэзДЬЌЯТЃЌИпTCгадЗЂгыМЬЗЂСНРрЃЌдЗЂЕФШчМвзхадИпЕЈЙЬДМбЊжЂЃЈЕЭУмЖШжЌЕААзЪмЬхШБЯнЃЉЁЂМвзхадapoBШБЯнжЂЁЂЖрдДадИпTCЁЂЛьКЯадИпжЌЕААзбЊжЂЁЃМЬЗЂЕФЖрМћгкЩіВЁзлКЯеїЁЂМззДЯйЙІФмМѕЭЫЁЂЬЧФђВЁЁЂШбЩяЕШЁЃЕЭTCбЊжЂвВгадЗЂадЕФгыМЬЗЂЕФЃЌЧАепШчМвзхадЕФЮоЛђЕЭІТжЌЕААзбЊжЂЃЌКѓепШчМзПКЁЂгЊбјВЛСМЁЂТ§адЯћКФадМВВЁЕШЁЃ
2.ВњЦЗУшЪі
гІВћЪіВњЦЗЫљВЩгУЕФММЪѕдРэЁЂжївЊдВФСЯЕФРДдДМАжЦБИЗНЗЈЁЂжївЊЩњВњЙЄвеЙ§ГЬЃЌзЂВсЩъЧыАќКЌжЪПиЦЗЁЂаЃзМЦЗЪБЃЌгІУїШЗжЦБИЗНЗЈМАЖЈжЕЃЈЫндДЃЉЧщПіЁЃ
3.гаЙиЩњЮяАВШЋадЗНУцЕФЫЕУї
ШчЪдМСжажївЊдВФСЯгЩИїжжЖЏЮяЁЂВЁдЬхЁЂШЫдДзщжЏКЭЬхвКЕШЩњЮяВФСЯОДІРэЛђЬэМгФГаЉЮяжЪжЦБИЖјГЩЃЌЮЊБЃжЄВњЦЗдкдЫЪфЁЂЪЙгУЙ§ГЬжаЖдЪЙгУепКЭЛЗОГЕФАВШЋЃЌбаОПепгІЬсЙЉЩЯЪідВФСЯгаЙиЩњЮяАВШЋадЕФЫЕУїЛђжЄУїЮФМўЁЃ
4.гаЙиВњЦЗжївЊбаОПНсЙћЕФзмНсКЭЦРМлЁЃ
5.ЦфЫћ
АќРЈЭЌРрВњЦЗдкЙњФкЭтХњзМЩЯЪаЕФЧщПіЃЌЯрЙиВњЦЗЫљВЩгУЕФММЪѕЗНЗЈМАСйДВгІгУЧщПіЃЌЩъЧызЂВсВњЦЗгыЙњФкЭтЭЌРрВњЦЗЕФвьЭЌЕШЁЃ
ЃЈЖўЃЉжївЊдВФСЯЕФбаОПзЪСЯЃЈШчашЬсЙЉЃЉ
АќРЈжївЊЗДгІГЩЗжЁЂжЪПиЦЗЁЂаЃзМЦЗЕШЕФбЁдёЁЂжЦБИМАЦфжЪСПБъзМЕФбаОПзЪСЯЃЛжЪПиЦЗгІЬсЙЉЯъЯИЕФЖЈжЕЪдбщзЪСЯЃЌаЃзМЦЗгІЬсЙЉЯъЯИЕФЫндДадЮФМўЃЌАќРЈОпЬхЫндДСДЁЂЪЕбщЗНЗЈЁЂЪ§ОнМАЭГМЦЗжЮіЕШЯъЯИзЪСЯЁЃ
ЃЈШ§ЃЉжївЊЩњВњЙЄвеКЭЗДгІЬхЯЕЕФбаОПзЪСЯЃЈШчашЬсЙЉЃЉ
1.жївЊЩњВњЙЄвеНщЩмЃЌПЩвдЭМБэЗНЪНБэЪОЃЛ
2.ШЗЖЈЗДгІЬхЯЕЃЈбљБОЁЂЪдМСЕШЃЉзюЪЪЬѕМўЕФбаОПзЪСЯЕШЁЃ
ЃЈЫФЃЉЗжЮіадФмЦРЙРзЪСЯ
ЩъЧыШЫгІЕБЬсНЛШ§ХњВњЦЗбажЦНзЖЮЖдЪдМСНјааЕФЫљгаадФмбщжЄбаОПзЪСЯЃЌЖдгкУПЯюЗжЮіадФмЕФЦРМлгІАќРЈОпЬхбаОПФПЕФЁЂЪЕбщЩшМЦЁЂбаОПЗНЗЈЁЂПЩНгЪмБъзМЁЂЪЕбщЪ§ОнЁЂЭГМЦЗНЗЈЕШЯъЯИзЪСЯЁЃгаЙиЗжЮіадФмбщжЄЕФБГОАаХЯЂвВгІдкЩъБЈзЪСЯжаЬхЯжЃЌАќРЈЪЕбщЕиЕуЁЂЪЪгУвЧЦїЁЂЪдМСЙцИёЁЂХњКХЁЂСйДВбљБОРДдДЕШЁЃ
ЖдгкзмЕЈЙЬДМВтЖЈЪдМСЃЌЗжЮіадФмгІАќРЈЪдМСПеАзЮќЙтЖШЁЂЯпадЗЖЮЇЁЂзМШЗЖШЁЂЗжЮіСщУєЖШЁЂОЋУмЖШЁЂЗжЮіЬивьадЁЂЦфЫћгАЯьМьВтЕФвђЫиЕШЁЃ
1.ЪдМСПеАзЮќЙтЖШ
гУжИЖЈПеАзбљЦЗЃЈШчДПЛЏЫЎЁЂЩњРэбЮЫЎЕШЃЉМгШыЙЄзїЪдМСзїЮЊбљЦЗЃЌдкЪдМСЙцЖЈжїВЈГЄЯТВтЪдЮќЙтЖШжЕЃЌжиИДСНДЮЃЌОљжЕМДЮЊПеАзЮќЙтЖШЁЃЫЋЪдМСаЭЪдМСгІВЛДѓгк0.080ЃЌЕЅЪдМСаЭЪдМСгІВЛДѓгк0.100ЁЃ
2.ЯпадЗЖЮЇ
2.1ЯпадЗЖЮЇЕФНЈСЂ
ЩъЧыШЫНЈСЂЪдМСЯпадЗЖЮЇЫљгУЕФбљБОЛљжЪгІОЁПЩФмгыСйДВЪЕМЪМьВтЕФбљБОЯрЫЦЃЌРэЯыЕФбљБОЮЊЗжЮіЮяХЈЖШДяЕНдЄЦкВтЖЈЩЯЯоЕФЛьКЯШЫдДбљБОЃЌжЦБИЕЭХЈЖШбљБОЪБгІГфЗжПМТЧЯЁЪЭЖдбљБОЛљжЪЕФгАЯьЁЃНЈСЂЯпадЗЖЮЇЪБЃЌашдкдЄЦкВтЖЈЗЖЮЇФкбЁдё7ЁЋ11ИіХЈЖШЫЎЦНЁЃР§ШчЃЌНЋдЄЦкВтЖЈЗЖЮЇМгПэжС130%ЃЌдкДЫЗЖЮЇФкбЁдёИќЖрЕФХЈЖШЫЎЦНЃЌШЛКѓвРОнЪЕбщНсЙћж№НЅМѕЩйЪ§ОнЕуЃЈзюжеВЛЕУЩйгк7ИіЫЎЦНЃЉжБжСБэЯжГіЯпадЙиЯЕЃЌПЩЗЂЯжзюПэЕФЯпадЗЖЮЇЁЃ
2.2ЯпадЗЖЮЇЕФбщжЄ
2.2.1гУНгНќЯпадЗЖЮЇЩЯЯоЕФИпХЈЖШбљБОКЭНгНќЯпадЗЖЮЇЯТЯоЕФЕЭХЈЖШбљБОЃЌЛьКЯГЩжСЩй5ИіЯЁЪЭХЈЖШЃЈxiЃЉЁЃЗжБ№ВтЪдбљБОЃЌУПИіЯЁЪЭХЈЖШВтЪд3ДЮЃЌЧѓГіУПИіЯЁЪЭХЈЖШВтЖЈНсЙћЕФОљжЕЃЈyiЃЉЁЃвдЯЁЪЭХЈЖШЃЈxiЃЉЮЊздБфСПЃЌвдВтЖЈНсЙћОљжЕЃЈyiЃЉЮЊвђБфСПЧѓГіЯпадЛиЙщЗНГЬЁЃМЦЫуЯпадЛиЙщЕФЯрЙиЯЕЪ§ЃЈrЃЉЃЌrЁн0.9900ЁЃ
2.2.2гУ2.2.1ЗНЗЈжаЯЁЪЭХЈЖШЃЈxiЃЉДњШыЧѓГіЯпадЛиЙщЗНГЬЃЌМЦЫуyiЕФЙРМЦжЕМАyiгыЙРМЦжЕЕФОјЖдЦЋВюЛђЯрЖдЦЋВюЃЌгІЗћКЯЩъЧыШЫИјЖЈжЕЁЃ
3.зМШЗЖШ
ВтЪдПЩгУгкЦРМлГЃЙцЗНЗЈЕФЙњМв/ЙњМЪБъзМЮяжЪ3ДЮЃЌВтЪдНсЙћМЧЮЊЃЈxiЃЉЃЌАДЙЋЪНЃЈ1ЃЉЗжБ№МЦЫуЯрЖдЦЋВюЃЈB%ЃЉЃЌШчЙћ3ДЮНсЙћЖМдкЁР10%ЗЖЮЇФкЃЌМДХаЮЊКЯИёЁЃШчЙћДѓгкЕШгк2ДЮЕФНсЙћВЛЗћКЯЃЌМДХаЮЊВЛКЯИёЁЃШчЙћга1ДЮНсЙћВЛЗћКЯЃЌдђгІжиаТСЌајВтЪд20ДЮЃЌВЂЗжБ№АДееЙЋЪНЃЈ1ЃЉМЦЫуЯрЖдЦЋВюЃЈB%ЃЉЃЌШчЙћДѓгкЕШгк19ДЮВтЪдЕФНсЙћЖМдкЁР10%ЗЖЮЇФкЃЌМДХаЮЊКЯИёЁЃ
B%ЃН|xiЃT|ЃЏTЁС100%ЁЁЁЁЁЁЁЁЁЁЁЁЃЈ1ЃЉ
ЪНжаЃК
xiЮЊВтЪдНсЙћЃЛ
TЮЊЙњМв/ЙњМЪБъзМЮяжЪБъЪОжЕЁЃ
4.ЗжЮіСщУєЖШ
ВтЪдИјЖЈХЈЖШЕФБЛВтЮяЪБЃЌдкЙцЖЈВЮЪ§ЯТЫљЕУЕФЮќЙтЖШВюжЕЃЈІЄAЃЉгІЗћКЯЩъЧыШЫИјЖЈЕФЗЖЮЇЁЃ
5.ОЋУмЖШ
5.1ХњФкОЋУмЖШ
жиИДВтЪдВЛЭЌХЈЖШЃЈШчИпЁЂжаЁЂЕЭЃЉЕФжЪПиЦЗЛђбљБО10ДЮЃЌЫљЕУНсЙћЕФБфвьЯЕЪ§ЃЈCVЃЉгІВЛДѓгк4.0%ЁЃ
5.2ХњМфВю
бЁШЁ3ИіВЛЭЌХњДЮЕФЪдМСЃЌВтЪдВЛЭЌХЈЖШЃЈШчИпЁЂжаЁЂЕЭЃЉЕФжЪПиЦЗЛђбљБОЃЌУПИіХњКХВтЖЈ3ДЮЃЌХњМфЯрЖдМЋВюЃЈRЃЉгІВЛДѓгк6.0%ЁЃ
6.ЗжЮіЬивьад
гІУїШЗвбжЊИЩШХвђЫиЖдВтЖЈНсЙћЕФгАЯьЃКЖдЕЈКьЫиЁЂИЪгЭШ§ѕЅЁЂбЊКьЕААзЁЂПЙЛЕбЊЫсЕШИЩШХЮяжЪЗжБ№НјаабщжЄЁЃЫЕУїбљБОЕФжЦБИЗНЗЈМАЪЕбщЕФЦРМлБъзМЃЌШЗЖЈИЩШХЮяжЪЕФНгЪмБъзМЁЃ
7.аЃзМЦЗЫндДМАжЪПиЦЗЖЈжЕЃЈШчЪЪгУЃЉ
аЃзМЦЗгІЬсЙЉЯъЯИЕФСПжЕЫндДзЪСЯЃЌАќРЈИГжЕЪдбщзЪСЯКЭЫндДадЮФМўЕШЃЛжЪПиЦЗгІЬсЙЉЯъЯИЕФЖЈжЕзЪСЯЁЃгІВЮееGB/T 21415?2008ЁЖЬхЭтеяЖЯвНСЦЦїаЕЩњЮябљЦЗжаСПЕФВтСПаЃзМЦЗКЭПижЦЮяжЪИГжЕЕФМЦСПбЇЫндДадЁЗЕФвЊЧѓЃЌЬсЙЉЦѓвЕЃЈЙЄзїЃЉаЃзМЦЗМАЪдМСКаХфЬзаЃзМЦЗИГжЕМАВЛШЗЖЈЖШМЦЫуМЧТМЃЌЬсЙЉжЪПиЦЗдкЫљгаЪЪгУЛњаЭЩЯЕФЖЈжЕМАЦфАажЕЗЖЮЇШЗЖЈЕФМЧТМЁЃ
8.ЦфЫћашзЂвтЮЪЬт
ЖдгкЪЪгУЖрИіЛњаЭЕФВњЦЗЃЌзЂВсЩъЧыЪБгІЬсЙЉШчВњЦЗЫЕУїЪщЁОЪЪгУЛњаЭЁПЯюжаЫљСаЕФЫљгааЭКХвЧЦїЕФадФмЦРЙРзЪСЯЁЃ
зЂВсЩъЧыжаАќКЌВЛЭЌЕФАќзАЙцИёЃЌашвЊЖдВЛЭЌАќзАЙцИёжЎМфЕФВювьНјааЗжЮіЛђбщжЄЃЌШчВЛЭЌАќзАЙцИёВњЦЗМфДцдкадФмВювьЃЌашвЊЬсНЛВЩгУУПИіАќзАЙцИёВњЦЗЕФЗжЮіадФмЦРЙРЁЃШчВЛЭЌАќзАЙцИёжЎМфВЛДцдкадФмВювьЃЌашвЊЬсНЛАќзАЙцИёжЎМфВЛДцдкадФмВювьЕФЯъЯИЫЕУїЃЌОпЬхЫЕУїВЛЭЌАќзАЙцИёжЎМфЕФВюБ№МАПЩФмВњЩњЕФгАЯьЁЃ
ЃЈЮхЃЉВЮПМЧјМфШЗЖЈзЪСЯ
гІУїШЗбаОПВЩгУЕФбљБОРДдДЁЂНЁПЕИіЬхЖЈвхЁЂЯъЯИЕФЪдбщзЪСЯЁЂЭГМЦЗНЗЈЕШЃЌВЮПМЧјМфПЩВЮПМЮФЯззЪСЯЃЌЕЋгІЕБЖджСЩй120Р§ЕФНЁПЕИіЬхНјаабщжЄЁЃ
ВњЦЗЫЕУїЪщЁОВЮПМЧјМфЁПЕФЯргІУшЪігІгыбаОПНсТлБЃГжвЛжТЁЃ
ЃЈСљЃЉЮШЖЈадбаОПзЪСЯ
1.ЮШЖЈадбаОПзЪСЯжївЊЩцМАСНВПЗжФкШнЃЌЩъБЈЪдМСЕФЮШЖЈадКЭЪЪгУбљБОЕФЮШЖЈадбаОПЁЃ
2.ЪдМСЕФЮШЖЈадАќРЈЪЕЪБЮШЖЈадЁЂдЫЪфЮШЖЈадЁЂПЊЦП/ИДШмЮШЖЈадЃЈШчЪЪгУЃЉЕШЃЌЩъЧыШЫгІжСЩйЬсЙЉ3ИіЩњВњХњДЮЕФЪЕЪБЮШЖЈадЁЂдЫЪфЮШЖЈадЁЂПЊЦП/ИДШмЮШЖЈадЃЈШчЪЪгУЃЉЕШбаОПзЪСЯЃЌАќРЈбаОПФПЕФЁЂЦРМлБъзМЁЂбаОПЗНЗЈКЭбаОПНсТлЕШЁЃ
3.ЪЪгУбљБОЕФЮШЖЈаджївЊАќРЈЪвЮТБЃДцЁЂРфВиКЭ/ЛђРфЖГЬѕМўЯТЕФгааЇЦкбщжЄЃЌдкКЯРэЮТЖШЗЖЮЇФкбЁдёЮТЖШЕуЃЌМфИєвЛЖЈЪБМфЖЮЖдДЂДцбљБОНјааадФмЕФЗжЮібщжЄЃЌДгЖјШЗШЯбљБОЕФаЇЦкЮШЖЈадЃЌвВПЩвдЭЈЙ§ЯрЙиЮФЯззЪСЯРДжЄУїЪЪгУбљБОЕФЮШЖЈадЁЃЪЪгУРфЖГБЃДцЕФбљБОгІЖдЖГШкДЮЪ§НјааЦРМлЁЃ
ЪдМСЮШЖЈадКЭбљБОЮШЖЈадСНВПЗжФкШнЕФбаОПНсЙћгІЗжБ№дкЫЕУїЪщЁОДЂДцЬѕМўМАгааЇЦкЁПКЭЁОбљБОвЊЧѓЁПСНЯюжаНјааЯъЯИЫЕУїЁЃ
ЃЈЦпЃЉСйДВЦРМлзЪСЯ
ДЫЯюФПвбОСаШыЁЖЙигкаТаоЖЉУтгкНјааСйДВЪдбщвНСЦЦїаЕФПТМЕФЭЈИцЁЗЃЈЙњМввЉЦЗМрЖНЙмРэОжЭЈИц2018ФъЕк94КХЃЉжаУтгкНјааСйДВЪдбщЕФЬхЭтеяЖЯЪдМСФПТМЁЃИљОнЬхЭтеяЖЯЪдМССйДВЦРМлЕФЯрЙивЊЧѓЃЌЩъЧыШЫПЩАДееЁЖУтгкНјааСйДВЪдбщЕФЬхЭтеяЖЯЪдМССйДВЦРМлзЪСЯЛљБОвЊЧѓЃЈЪдааЃЉЁЗЃЈЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЭЈИц2017ФъЕк179КХЃЉвЊЧѓНјааСйДВЦРМлЁЃШчЮоЗЈАДвЊЧѓНјааСйДВЦРМлЃЌгІНјааСйДВЪдбщЃЌСйДВЪдбщЕФПЊеЙЁЂЗНАИЕФжЦЖЈвдМАБЈИцЕФзЋаДЕШОљгІЗћКЯЯрЙиЗЈЙцМАЁЖЬхЭтеяЖЯЪдМССйДВЪдбщММЪѕжИЕМддђЁЗЕФвЊЧѓЁЃ
1.СйДВЦРМлЭООЖ
СйДВЦРМлЪЧжИЩъЧыШЫЭЈЙ§ЖдКИЧдЄЦкгУЭОМАИЩШХвђЫиЕФСйДВбљБОЕФЦРЙРЁЂзлКЯЮФЯззЪСЯЕШЗЧСйДВЪдбщЕФЗНЪНЖдВњЦЗСйДВадФмНјааЦРМлЕФЙ§ГЬЁЃ
ЩъЧыШЫПЩздааЛђепЮЏЭаОГФкЛњЙЙЛђЪЕбщЪвЭъГЩИУВњЦЗЕФСйДВЦРМлЃЌЪдбщЕиЕуЕФЩшЪЉЁЂЩшБИЁЂЛЗОГЕШгІФмЙЛТњзуИУВњЦЗЕФМьВтвЊЧѓЁЃЩъЧыШЫЖдећИіЪдбщЙ§ГЬНјааЙмРэКЭЪдбщЪ§ОнЕФецЪЕадНјааИКд№ЁЃСйДВЦРМлБЈИцзїЮЊСйДВЦРМлзЪСЯдкзЂВсЪБЬсНЛЃЌЦфЫћСйДВЦРМлЯрЙизЪСЯШчЪдбщЗНАИЁЂдЪММЧТМЕШгЩЩъЧыШЫНјааБЃЙмЃЌБЃЙмЦкЯожСЩйЮЊ10ФъЁЃОГЭтЩъЧыШЫПЩЭЈЙ§ЦфдкжаЙњОГФкЕФДњРэШЫЃЌПЊеЙЯрЙиСйДВЦРМлЙЄзїЁЃвдЯТМђвЊЫЕУїСйДВЦРМлЕФЯрЙиФкШнЁЃ
1.1баОПЗНЗЈ
змЕЈЙЬДМВтЖЈЪдМСКаФПЧАвбгаЯргІЕФВЮПМЗНЗЈЃЈALBKЗЈЁЂЩЋЦзЗЈЕШЃЉКЭОГФквбЩЯЪаЭЌРрВњЦЗЃЌЩъЧыШЫПЩвдбЁдёЦфжавЛжжЗНЗЈПЊеЙБШНЯбаОПЪдбщЁЃ
гыВЮПМЗНЗЈНјааБШНЯбаОПЪдбщЃЌПМВьД§ЦРМлЪдМСгыВЮПМЗНЗЈЕФвЛжТадЁЃвЛжТадЦРМлгІбЁдёдкВЮПМЪЕбщЪвПЊеЙбаОПЃЌВЮПМЪЕбщЪвгІОпгажаЙњКЯИёЦРЖЈЙњМвШЯПЩЮЏдБЛсШЯПЩЙигкИУЯюФПЕФМьВтзЪжЪЁЃ
гыОГФквбЩЯЪаЭЌРрВњЦЗНјааБШНЯбаОПЪдбщЃЌжЄУїСНепОпгаЕШаЇадЁЃгІбЁдёФПЧАСйДВЦеБщШЯЮЊжЪСПНЯКУЕФВњЦЗзїЮЊЖдБШЪдМСЃЌЭЌЪБгІГфЗжСЫНтЖдБШЪдМСЕФаХЯЂЃЌАќРЈЗНЗЈбЇЁЂдЄЦкгУЭОЁЂжївЊадФмжИБъЁЂаЃзМЦЗЕФЫндДЧщПівдМАВЮПМЧјМфЕШЃЌВЂЬсЙЉвбЩЯЪаВњЦЗЕФОГФкзЂВсаХЯЂМАЫЕУїЪщЁЃ
1.2ЪдбщЩшМЦ
ЩъЧыШЫгІИљОнЩъЧызЂВсВњЦЗЕФЬиадНЈСЂОпЬхЕФЪдбщЗНАИЃЌВЂЯъЯИЫЕУїЖдБШЪдМС/ЗНЗЈЕФбЁдёЁЂШыХХБъзМЁЂбљБОСПвЊЧѓвдМАОпЬхЪдбщВйзїЙ§ГЬЁЂЪдбщжмЦкЁЂЩшУЄвЊЧѓЁЂЭГМЦЗжЮіЗНЗЈЕШФкШнЁЃ
1.2.1бљБОвЊЧѓ
ЦРМлгУбљБОгІРДдДгкШЫЬхбљБОЃЌбљБОЕФРДдДПЩзЗЫнЁЃЦРМлгУбљБОгІКИЧЩъЧызЂВсВњЦЗдЄЦкгУЭОКЭКЌгаЕЈКьЫиЁЂбЊКьЕААзЁЂИЪгЭШ§ѕЅЁЂПЙЛЕбЊЫсЕШИЩШХЮяжЪЕФбљБОЃЌВЂгІПМТЧЕНВЛЭЌШЫШКжЎМфЕФВювьЕШЁЃЫљгУбљБОгІФмЙЛГфЗжЕФБЛгУгкЦРМлВњЦЗСйДВЪЙгУЕФАВШЋадЁЂгааЇадЁЃ
бљБОЪ§СПгІВЩгУКЯРэЕФЭГМЦбЇЗНЗЈЃЈШчФПБъжЕЗЈЕШЃЉНјааМЦЫуЃЌВЂЗћКЯЭГМЦбЇвЊЧѓЁЃПЩбЁдёзмбљБОСПВЛЩйгк40Ч€ЗжБ№ВЩгУД§ЦРМлЪдМСКЭЖдБШЪдМС/ВЮПМЗНЗЈНјааЫЋЗнВтЖЈЕФЗНЪНЃЌврПЩбЁдёзмбљБОСПВЛЩйгк100Ч€ЗжБ№ВЩгУД§ЦРМлЪдМСКЭЖдБШЪдМС/ВЮПМЗНЗЈНјааЕЅДЮВтЖЈЕФЗНЪНЁЃЦфжаВЮПМЧјМфвдЭтбљБОгІВЛЩйгк50%ЃЌЭЌЪБгІзЂживНбЇОіЖЈЫЎЦНЃЈ6.2mmol/LЃЉИННќбљБОЕФбЁдёЃЌВЂОЁСПКИЧМьВтЗЖЮЇЁЃ
ЪдбщЧАгІЩшЖЈСйДВЦРМладФмжИБъЕФПЩНгЪмБъзМ,ШчЙћБШНЯбаОПЪдбщНсЙћЮоЗЈДяЕНдЄЩшБъзМЃЌдђгІЪЪЕБРЉДѓбљБОСПНјааЦРМлЁЃ
ЦРМлгУЕФбљБОРраЭгІгызЂВсЩъЧыБЃГжвЛжТЁЃЖдгкОпгаПЩБШадЕФВЛЭЌбљБОРраЭЃЌШчбЊЧхКЭбЊНЌбљБОЃЌПЩдкЗжЮіадФмЦРЙРжаЖдбљБОЪЪгУадНјаабаОПЃЌЛђдкСйДВЦРМлжаЖдУПжжбљБОРраЭЗжБ№НјааЗћКЯЭГМЦбЇвтвхЪ§СПЕФЦРЙРЁЃ
1.2.2ЪдбщВйзїМАжмЦк
ЩъЧыШЫПЩвдЭЈЙ§ЖдзмбљБОСПВЛЩйгк40Р§ЃЌгУД§ЦРМлЪдМСКЭЖдБШЪдМС/ВЮПМЗНЗЈНјааЦНааЪдбщЃЌЖдУПЗнСйДВбљБОзїЫЋЗнВтЖЈЁЃдкЪдбщВйзїЙ§ГЬжагІЖдбљБОНјааБрУЄЃЌЪдбщМьВтжмЦкжСЩйЮЊ5ЬьЁЃ
ЩъЧыШЫвВПЩвдЭЈЙ§ЖдзмбљБОСПВЛЩйгк100Р§ЃЌгУД§ЦРМлЪдМСКЭЖдБШЪдМС/ВЮПМЗНЗЈНјааЦНааЪдбщЃЌзїЕЅДЮВтЖЈЁЃдкЪдбщВйзїЙ§ГЬжагІЖдбљБОНјааБрУЄЃЌЪдбщМьВтжмЦкжСЩйЮЊ5ЬьЁЃ
1.2.3Ъ§ОнЪеМЏКЭДІРэ
гІЪзЯШНјааРыШКжЕЙлВьЃЌРыШКжЕЕФИіЪ§ВЛЕУГЌЙ§ЯожЕЁЃШєЮДГЌЯоЃЌПЩЩОГ§РыШКжЕКѓНјааЗжЮіЃЛШєГЌГіЯожЕЃЌдђашКЯРэЗжЮідвђВЂПМТЧОРе§ДыЪЉЃЌБивЊЪБжиаТЪеМЏбљБОНјааЗжЮіЁЃРыШКжЕЗжЮіКЭДІРэЗНЗЈгІгавРОнЁЃ
1.2.4ЭГМЦбЇЗжЮі
ЖдЪдбщНсЙћЕФЭГМЦгІбЁдёКЯЪЪЕФЭГМЦЗНЗЈЃЌШчЯрЙиЗжЮіЁЂЯпадЛиЙщЁЂЦЋва/ЦЋВюЗжЮіЕШЁЃЖдгкЖдБШЪдбщЕФЕШаЇадбаОПЃЌзюГЃгУЪЧЖдД§ЦРЪдМСКЭЖдБШЪдМС/ВЮПМЗНЗЈСНзщМьВтНсЙћЕФЯрЙиМАЯпадЛиЙщЗжЮіЃЌгІжиЕуЙлВьЯрЙиЯЕЪ§ЃЈrжЕЃЉЛђХаЖЈЯЕЪ§ЃЈR2ЃЉЁЂЛиЙщФтКЯЗНГЬЃЈаБТЪКЭyжсНиОрЃЉЕШжИБъЁЃ
1.3СйДВЦРМлБЈИц
СйДВЦРМлБЈИцгІЖдЪдбщЩшМЦЁЂЪдбщЪЕЪЉЧщПіКЭЪ§ОнЗжЮіЗНЗЈЕШНјааЧхЮњЕФУшЪіЁЃгІжСЩйАќРЈШчЯТФкШнЃК
1.3.1ЛљБОаХЯЂЃЌШчВњЦЗУћГЦЁЂЩъЧыШЫУћГЦМАСЊЯЕЗНЪНЁЂЪдбщЪБМфМАЕиЕуЕШЁЃ
1.3.2ЪдбщЩшМЦЃЌЯъЯИЫЕУїЖдБШЪдМС/ЗНЗЈбЁдёЁЂбљБОШызщКЭХХГ§БъзМЁЂбљБОСПвЊЧѓЁЂЩшУЄвЊЧѓЁЂЭГМЦЗжЮіЗНЗЈЕФбЁдёЕШФкШнЁЃ
1.3.3ЪдбщЪЕЪЉЧщПіЃЌОпЬхАќРЈЃК
бљБОбЁдёЧщПіЃЌАќРЈР§Ъ§ЁЂбљБОЗжВМЕШЁЃбљБОР§Ъ§гІЯъЯИЫЕУїМЦЫуЗНЗЈМАвРОнЁЃСйДВЦРМлЫљгУВњЦЗаХЯЂЃЌШчЦРМлгУЪдМСЁЂЖдБШЪдМС/ЗНЗЈЁЂХфКЯЪЙгУЕФЦфЫћЪдМС/вЧЦїЕФВњЦЗУћГЦЁЂЩњВњЦѓвЕЁЂЙцИё/аЭКХЁЂХњКХЕШЁЃЪЕбщЙ§ГЬУшЪіЁЃЪдбщЙмРэЃЌАќРЈВЮМгШЫдБЁЂжЪСППижЦЧщПіЁЂЪ§ОнЙмРэЁЂГіЯжЕФЮЪЬтМАДІРэДыЪЉЕШЁЃЪ§ОнЗжЮіМАЦРМлНсЙћзмНсЃЌИљОнШЗЖЈЕФЭГМЦЗНЗЈЖдМьВтЪ§ОнНјааЭГМЦЗжЮіЃЌЖдВњЦЗЕФСйДВадФмНјааКЯРэЦРМлЁЃЦРМлЪ§ОнБэгІвдИНМўаЮЪНЖдШызщЕФбљБОЧщПіНјааЛузмУшЪіЃЌгІжСЩйАќРЈЃКПЩЫндДбљБОБрКХЁЂбљБОЛљБОаХЯЂЁЂбљБОРраЭЁЂЦРМлгУЪдМСКЭЖдБШЪдМС/ЗНЗЈМьВтНсЙћЁЂбљБОСйДВБГОАаХЯЂЕШЁЃ
ЦРМлБЈИцгІгЩЩъЧыШЫ/ДњРэШЫЧЉеТЁЃ
1.4ЮФЯззЪСЯ
Г§вдЩЯСйДВЦРМлБЈИцЭт,ЖдФтЩъБЈВњЦЗСйДВадФмНјааЦРМлЕФЯрЙиЮФЯз,ПЩзїЮЊВЙГфСйДВЦРМлзЪСЯЬсНЛЁЃЮФЯзЕФМьЫїЁЂЩИбЁКЭЗжЮіВЮееЁЖвНСЦЦїаЕСйДВЦРМлММЪѕжИЕМддђЁЗЕФЮФЯзМьЫївЊЧѓЁЃ
2.СйДВЪдбщЭООЖ
СйДВЪдбщЕФПЊеЙЁЂЗНАИЕФжЦЖЈвдМАБЈИцЕФзЋаДЕШОљгІЗћКЯЯрЙиЗЈЙцМАЁЖЬхЭтеяЖЯЪдМССйДВЪдбщММЪѕжИЕМддђЁЗЕФвЊЧѓЁЃ
ЃЈАЫЃЉВњЦЗЗчЯеЗжЮізЪСЯ
ЖдЬхЭтеяЖЯЪдМСВњЦЗЪйУќжмЦкЕФИїИіЛЗНк,ДгдЄЦкгУЭОЁЂПЩФмЕФЪЙгУДэЮѓЁЂгыАВШЋадгаЙиЕФЬиеїЁЂвбжЊКЭПЩдЄМћЕФЮЃКІЕШЗНУцЕФХаЖЈвдМАЖдЛМепЗчЯеЕФЙРМЦНјааЗчЯеЗжЮіЁЂЗчЯеЦРМлКЭЯргІЕФЗчЯеПижЦЛљДЁЩЯЃЌаЮГЩЗчЯеЙмРэБЈИцЃЌгІЕБЗћКЯЯрЙиYY/T 0316-2016аавЕБъзМЕФвЊЧѓЁЃ
ЗчЯеЗжЮігІАќКЌЕЋВЛНіЯогквдЯТЗНУцЕФФкШнЃКдЄЦкгУЭОДэЮѓАќРЈЃКЩшМЦПЊЪМЪБЮДЩшЖЈдЄЦкЗжЮіЮяЁЂЮДзїЪЪгУЛњаЭбщжЄЁЂЮДеыЖдЬиЖЈЕФбљБОРраЭбщжЄЁЃадФмЬиеїЪЇаЇАќРЈЃКОЋУмЖШЪЇаЇЁЂзМШЗЖШЪЇаЇЁЂЗЧЬивьадЁЂЮШЖЈадЪЇаЇЁЂВтСПЗЖЮЇЪЇаЇЁЂЖЈСПЪЇаЇЁЂСПжЕЫндДЪЇаЇЁЂаЃзМЪЇаЇЁЃВЛе§ШЗЕФНсЙћАќРЈЃКХфЗНДэЮѓЁЂВЩЙКЕФдСЯЮДФмДяЕНЩшМЦвЊЧѓЕФадФмЁЂдВФСЯДЂДцЬѕМўВЛе§ШЗЁЂЪЙгУСЫЙ§ЦкЕФдВФСЯЁЂЗДгІЬхЯЕВЛе§ШЗЁЂЪдМСгыАќзАВФСЯВЛЯрШнЁЃПЩФмЕФЪЙгУДэЮѓАќРЈЃКЩњВњепЮДАДееЩњВњСїГЬВйзїЃЌМьбщепЮДАДеедСЯЁЂАыГЩЦЗЁЂГЩЦЗМьбщБъзМВйзїЃЌзАХфЙ§ГЬзщЗнЁЂБъЧЉЁЂЫЕУїЪщЕШТЉзАЛђЮѓзАЃЌГЩЦЗДЂДцЛђдЫЪфВЛЕБЃЌПЭЛЇЮДВЮееВњЦЗЫЕУїЪщЩшжУВЮЪ§ЛђЪЙгУЁЃгыАВШЋадгаЙиЕФЬиеїАќРЈЃКгаЖОЛЏбЇЪдМСЕФЛЏбЇЮлШОЁЂбљБОЕФЧБдкЩњЮяЮлШОЁЂВЛПЩЛиЪеАќзАЛђЫмСЯЕФЛЗОГЮлШОЁЃ
ЃЈОХЃЉВњЦЗММЪѕвЊЧѓ
ЩъЧыШЫгІЕБдкдВФСЯжЪСПКЭЩњВњЙЄвеЮШЖЈЕФЧАЬсЯТЃЌИљОнВњЦЗЧАЦкбаОПЕФНсЙћЃЌвРОнЙњМвБъзМЁЂаавЕБъзММАгаЙиЮФЯзЃЌАДееЁЖЙигкЗЂВМвНСЦЦїаЕВњЦЗММЪѕвЊЧѓБраДжИЕМддђЕФЭЈИцЁЗЃЈЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЭЈИц2014ФъЕк9КХЃЉЕФгаЙивЊЧѓЃЌФтЖЈВњЦЗММЪѕвЊЧѓЃЌФкШнжївЊАќКЌВњЦЗадФмжИБъКЭМьбщЗНЗЈЁЃ
ЯТУцОЭВњЦЗММЪѕвЊЧѓжаЩцМАЕФВњЦЗЪЪгУЕФЯрЙиБъзМКЭжївЊадФмжИБъЕШЯрЙиФкШнзїМђвЊа№ЪіЁЃ
1.ВњЦЗЪЪгУЕФЯрЙиБъзМЃК
Бэ1ЯрЙиВњЦЗБъзМ
|
GB/T
21415-2008
|
ЁЖЬхЭтеяЖЯвНСЦЦїаЕЩњЮябљЦЗжаСПЕФВтСПаЃзМЦЗКЭПижЦЮяжЪИГжЕЕФМЦСПбЇЫндДадЁЗ
|
|
GB/T
26124-2011
|
ЁЖСйДВЛЏбЇЬхЭтеяЖЯЪдМСЃЈКаЃЉЁЗ
|
|
YY/T
1206-2013
|
ЁЖзмЕЈЙЬДМВтЖЈЪдМСКаЃЈбѕЛЏУИЗЈЃЉЁЗ
|
|
WS/T362-2011
|
ЁЖбЊЧхЕЈЙЬДМВЮПМВтСПГЬађЗжЙтЙтЖШЗЈЁЗ
|
|
WS/T
120-1999
|
ЁЖбЊЧхзмЕЈЙЬДМЕФУИЗЈВтЖЈЁЗ
|
2.жївЊадФмжИБъЃК
2.1ЭтЙл
ЗћКЯЩъЧыШЫЙцЖЈЕФЭтЙлвЊЧѓЁЃ
2.2зАСП
вКЬхЪдМСЕФзАСПгІВЛЩйгкБъЪОСПЁЃ
2.3ЪдМСПеАзЮќЙтЖШ
гУжИЖЈПеАзбљЦЗЃЈШчДПЛЏЫЎЁЂЩњРэбЮЫЎЕШЃЉМгШыЙЄзїЪдМСзїЮЊбљЦЗЃЌдкЪдМСКаЙцЖЈжїВЈГЄЯТВтЪдЮќЙтЖШжЕЃЌжиИДСНДЮЃЌОљжЕМДЮЊПеАзЮќЙтЖШЁЃЫЋЪдМСаЭЪдМСКагІВЛДѓгк0.080ЃЌЕЅЪдМСаЭЪдМСКагІВЛДѓгк0.100ЁЃ
2.4ЯпадЗЖЮЇ
ЯпадЗЖЮЇгІжСЩйАќКЌ2ЁЋ10mmol/LЁЃ
2.4.1ЯрЙиЯЕЪ§ЃЈrЃЉ
ЯпадЯрЙиЯЕЪ§rгІВЛаЁгк0.9900ЁЃ
2.4.2ЯпадЦЋВю
ЯпадЦЋВюгІВЛГЌЙ§ЩъЧыШЫИјЖЈжЕЁЃЯпадЦЋВюПЩЗжЖЮУшЪіЛђСЌајУшЪіЃЌШєЗжЖЮУшЪіЃЌгІзЂвтЗжЖЮЕуЕФШЗЖЈЁЃ
2.5зМШЗЖШ
ЪдМСКаВтЪдПЩгУгкЦРМлГЃЙцЗНЗЈЕФЙњМв/ЙњМЪБъзМЮяжЪ3ДЮЃЌВтЪдНсЙћМЧЮЊЃЈxiЃЉЃЌАДЙЋЪНЃЈ1ЃЉЗжБ№МЦЫуЯрЖдЦЋВюЃЈB%ЃЉЃЌШчЙћ3ДЮНсЙћЖМдкЁР10%ЗЖЮЇФкЃЌМДХаЮЊКЯИёЁЃШчЙћДѓгкЕШгк2ДЮЕФНсЙћВЛЗћКЯЃЌМДХаЮЊВЛКЯИёЁЃШчЙћга1ДЮНсЙћВЛЗћКЯЃЌдђгІжиаТСЌајВтЪд20ДЮЃЌВЂЗжБ№АДееЙЋЪНЃЈ1ЃЉМЦЫуЯрЖдЦЋВюЃЈB%ЃЉЃЌШчЙћДѓгкЕШгк19ДЮВтЪдЕФНсЙћЖМдкЁР10%ЗЖЮЇФкЃЌМДХаЮЊКЯИёЁЃ
B%ЃН|xiЃT|ЃЏTЁС100%ЁЁЁЁЁЁЁЁЁЁЁЁЃЈ1ЃЉ
ЪНжаЃК
xiЮЊВтЪдНсЙћЃЛ
TЮЊЙњМв/ЙњМЪБъзМЮяжЪБъЪОжЕЁЃ
2.6ЗжЮіСщУєЖШ
ЪдМСКаВтЪдИјЖЈХЈЖШЕФБЛВтЮяЪБЃЌдкЪдМСКаЙцЖЈВЮЪ§ЯТЫљЕУЕФЮќЙтЖШВюжЕЃЈІЄAЃЉгІЗћКЯЩъЧыШЫИјЖЈЕФЗЖЮЇЁЃ
2.7ОЋУмЖШ
2.7.1ХњФкОЋУмЖШ
жиИДВтЪдВЛЭЌХЈЖШЃЈШчИпЁЂжаЁЂЕЭЃЉЕФжЪПиЦЗЛђбљБО10ДЮЃЌЫљЕУНсЙћЕФБфвьЯЕЪ§ЃЈCVЃЉгІВЛДѓгк4.0%ЁЃ
2.7.2ХњМфВю
бЁШЁ3ИіВЛЭЌХњДЮЕФЪдМСКаЃЌВтЪдВЛЭЌХЈЖШЃЈШчИпЁЂжаЁЂЕЭЃЉЕФжЪПиЦЗЛђбљБОЃЌУПИіХњКХВтЖЈ3ДЮЃЌЪдМСКаХњМфЯрЖдМЋВюЃЈRЃЉгІВЛДѓгк6.0%ЁЃ
2.8ЮШЖЈад
2.8.1аЇЦкФЉЮШЖЈад
ЪдМСКадкЙцЖЈЕФБЃДцЬѕМўЯТБЃДцжСгааЇЦкФЉНјааМьбщЃЌВњЦЗадФмгІЗћКЯ2.1ЁЂ2.3ЁЂ2.4ЁЂ2.5ЁЂ2.6ЁЂ2.7.1ЕФвЊЧѓЁЃ
2.8.2МгЫйЮШЖЈад
ИљОнЪдМСКаЕФгааЇЦкЃЌНЋЪдМСКаЗХжУгк37ЁцЬѕМўЯТЃЌвЛЖЈЪБМфЃЈЭЈГЃЪЧ3dЁЋ7dЃЉКѓНјааМьбщЃЌВњЦЗадФмгІЗћКЯ2.1ЁЂ2.3ЁЂ2.4ЁЂ2.5ЁЂ2.6ЁЂ2.7.1ЕФвЊЧѓЁЃ
зЂ1ЃКШШЮШЖЈадВЛФмгУгкЭЦЕМВњЦЗгааЇЦкЃЌГ§ЗЧЪЧВЩгУЛљгкДѓСПЕФЮШЖЈадбаОПЪ§ОнНЈСЂЕФЭЦЕМЙЋЪНЁЃ
зЂ2ЃКИљОнВњЦЗЬиадПЩбЁдёвдЩЯЗНЗЈжЎвЛНјаабщжЄЃЌЕЋЫљбЁгУЗНЗЈвЫФмбщжЄВњЦЗЕФЮШЖЈадЃЌвдБЃжЄдкаЇЦкФкВњЦЗадФмЗћКЯВњЦЗММЪѕвЊЧѓЕФЙцЖЈЁЃ
2.9аЃзМЦЗКЭжЪПиЦЗЃЈШчЪЪгУЃЉ
гІАќКЌЭтЙлЁЂзАСПЃЈШчЪЪгУЃЉЁЂзМШЗЖШЁЂОљвЛадЁЂЮШЖЈадЁЃ
2.9.1 ЭтЙл
ЗћКЯЩъЧыШЫЙцЖЈЕФЭтЙлвЊЧѓЁЃ
2.9.2зАСП
вКЬхаЃзМЦЗКЭжЪПиЦЗЕФзАСПгІВЛЩйгкБъЪОСПЁЃ
2.9.3зМШЗЖШ
2.9.3.1аЃзМЦЗзМШЗЖШ
ЩъЧыШЫПЩВЩгУЙњМв/ЙњМЪБъзМЮяжЪ/ОпгаЫндДадЕФЙЄзїаЃзМЦЗзїаЃзМЧњЯпКѓВтЖЈаЃзМЦЗЕФЗНЗЈЃЌМЦЫуВтЪдНсЙћгыБъЪОжЕЕФЯрЖдЦЋВюЃЌЦфНсЙћгІдкЁР10.0%ЗЖЮЇФкЁЃ
2.9.3.2 жЪПиЦЗзМШЗЖШ
ВтЖЈЖЈжЕжЪПиЦЗЃЌВтЪдНсЙћгІдкЩъЧыШЫИјЖЈЕФЗЖЮЇФкЁЃ
2.9.4ОљвЛад
ШЁЯрЭЌХњКХЕФаЃзМЦЗЛђжЪПиЦЗ10ЦПЃЌУПЦПВтЪд1ДЮЃЌАДЙЋЪНЃЈ2ЃЉКЭЙЋЪНЃЈ3ЃЉМЦЫуВтЪдНсЙћЕФЦНОљжЕЃЈ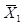 ЃЉКЭБъзМВюS1ЃЛСэгУЩЯЪіаЃзМЦЗЛђжЪПиЦЗжаЕФ1ЦПСЌајВт10ДЮЃЌМЦЫуВтЪдНсЙћЕФЦНОљжЕЃЈ
ЃЉКЭБъзМВюS1ЃЛСэгУЩЯЪіаЃзМЦЗЛђжЪПиЦЗжаЕФ1ЦПСЌајВт10ДЮЃЌМЦЫуВтЪдНсЙћЕФЦНОљжЕЃЈ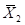 ЃЉКЭБъзМВюS2ЃЛдйАДЙЋЪН(4)КЭЙЋЪН(5)МЦЫуSЦПМфКЭCVЦПМфЃЌЦфНсЙћгІЗћКЯЩъЧыШЫЕФЙцЖЈЁЃ
ЃЉКЭБъзМВюS2ЃЛдйАДЙЋЪН(4)КЭЙЋЪН(5)МЦЫуSЦПМфКЭCVЦПМфЃЌЦфНсЙћгІЗћКЯЩъЧыШЫЕФЙцЖЈЁЃ
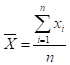 ЁЁЁЁЁЁЁЁЁЁЁЁЁЁЃЈ2ЃЉ
ЁЁЁЁЁЁЁЁЁЁЁЁЁЁЃЈ2ЃЉ
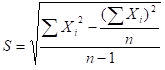 ЁЁЁЁЁЁЁЁЁЃЈ3ЃЉ
ЁЁЁЁЁЁЁЁЁЃЈ3ЃЉ
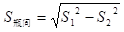 ЁЁЁЁЁЁЁЁЁЁЁЃЈ4ЃЉ
ЁЁЁЁЁЁЁЁЁЁЁЃЈ4ЃЉ
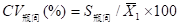 ЁЁЁЁЁЁЁЁЁЃЈ5ЃЉ
ЁЁЁЁЁЁЁЁЁЃЈ5ЃЉ
ЕБS1<S2ЪБЃЌСюCVЦПМф=0
ЪНжаЃК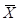 ----ЦНОљжЕЃЛS----БъзМВюЃЛ
----ЦНОљжЕЃЛS----БъзМВюЃЛ
n----ВтСПДЮЪ§ЃЛxi----жИЖЈВЮЪ§Екi ДЮВтСПжЕЁЃ
2.10ЮШЖЈад
аЃзМЦЗЁЂжЪПиЦЗдкЙцЖЈЕФБЃДцЬѕМўЯТБЃДцжСгааЇЦкФЉНјааМьбщЃЌадФмгІЗћКЯзМШЗЖШКЭОљвЛадЕФвЊЧѓЁЃ
ЃЈЪЎЃЉВњЦЗзЂВсМьбщБЈИц
ОпгаЯргІвНСЦЦїаЕМьбщзЪжЪКЭГаМьЗЖЮЇЕФвНСЦЦїаЕМьбщЛњЙЙГіОпЕФзЂВсМьбщБЈИцКЭВњЦЗММЪѕвЊЧѓдЄЦРМлвтМћЁЃзмЕЈЙЬДМЯюФПФПЧАгаЙњМв/ЙњМЪБъзМЮяжЪЃЌНЈвщЪЙгУЙњМв/ЙњМЪБъзМЮяжЪНјаазЂВсМьбщЁЃ
ЃЈЪЎвЛЃЉВњЦЗЫЕУїЪщ
ЫЕУїЪщГадиСЫВњЦЗдЄЦкгУЭОЁЂМьбщЗНЗЈЁЂМьбщНсЙћЕФНтЪЭвдМАзЂвтЪТЯюЕШживЊаХЯЂЃЌЪЧжИЕМЪЕбщЪвЙЄзїШЫдБе§ШЗВйзїЁЂСйДВвНЩњеыЖдМьбщНсЙћИјГіКЯРэвНбЇНтЪЭЕФживЊвРОнЃЌвђДЫЃЌВњЦЗЫЕУїЪщЪЧЬхЭтеяЖЯЪдМСзЂВсЩъБЈзюживЊЕФЮФМўжЎвЛЁЃ
НсКЯЁЖЙигкЗЂВМЬхЭтеяЖЯЪдМСЫЕУїЪщБраДжИЕМддђЕФЭЈИцЁЗЃЈЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЭЈИц2014ФъЕк17КХЃЉЕФвЊЧѓЃЌЯТУцЖдзмЕЈЙЬДМВтЖЈЪдМСКаЫЕУїЪщЕФжиЕуФкШнНјааЯъЯИЫЕУїЁЃ
1.ЁОВњЦЗУћГЦЁП
ЪдМСКаУћГЦгЩШ§ВПЗжзщГЩЃКБЛВтЮяжЪЕФУћГЦЁЂгУЭОЁЂЗНЗЈЛђдРэЁЃШчЃКзмЕЈЙЬДМВтЖЈЪдМСКаЃЈCHOD-PAPЗЈЃЉЁЃ
2.ЁОАќзАЙцИёЁП
2.1АќзАЙцИёгІУїШЗЕЅЁЂЫЋЛђЦфЫћЖрЪдМСРраЭЁЃ
2.2АќзАЙцИёгІзЂУїПЩВтЪдЕФбљБОЪ§ЛђзАСПЃЌШчxxВтЪд/КаЁЂxxmLЁЃ
2.3ШчгаЛѕКХЃЌПЩдіМгЛѕКХаХЯЂЁЃ
3.ЁОдЄЦкгУЭОЁП
гІжСЩйАќРЈвдЯТФкШнЃК
3.1ЫЕУїЪдМСКагУгкЬхЭтЖЈСПВтЖЈШЫбЊЧхЁЂбЊНЌЕШбљБОжазмЕЈЙЬДМЕФКЌСПЁЃ
3.2УїШЗгызмЕЈЙЬДМВтЖЈЯрЙиЕФСйДВЪЪгІжЂБГОАЧщПіЁЃ
4.ЁОМьбщдРэЁП
БъБОжаЕФЕЈЙЬДМѕЅБЛЕЈЙЬДМѕЅУИЃЈCHERЃЉЫЎНтЮЊгЮРыЕЈЙЬДМЃЌКЭБъБОжаДцдкЕФгЮРыЕЈЙЬДМвЛЦ№БЛЕЈЙЬДМбѕЛЏУИЃЈCHODЃЉбѕЛЏЮЊЕЈчоЯЉЭЊКЭЙ§бѕЛЏЧтЃЌКѓепдйОЙ§бѕЛЏЮяУИЃЈPODЃЉДпЛЏЃЌЪЙ4-АБЛљАВЬцБШСжКЭЗгЗЂЩњбѕЛЏЫѕКЯЗДгІЃЌЩњГЩКьЩЋѕЋбЧАЗ(ГЦTrinderЗДгІЃЌЛђГЦPAPЗДгІ)ЁЃБъБОжаЕФЕЈЙЬДМКЌСПКЭЩњГЩЕФѕЋбЧАЗГЪе§БШЃЌ500nmзѓгвВЈГЄМьВтѕЋбЧАЗЕФЮќЙтЖШЃЌЖдееаЃзМЮяМДПЩМЦЫуГіБъБОжаЕФзмЕЈЙЬДМКЌСПЃЌЦфЗДгІЪНШчЯТЃК
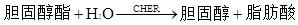
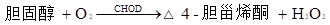
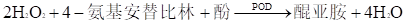
5.ЁОжївЊзщГЩГЩЗжЁП
гІУїШЗвдЯТФкШнЃК
5.1ЫЕУїЪдМСКаАќКЌзщЗжЕФУћГЦЁЂБШР§ЛђХЈЖШЃЛЖдгкЖрзщЗжЪдМСКаЃЌгІУїШЗВЛЭЌХњДЮЪдМСжаИїзщЗжЪЧЗёПЩвдЛЅЛЛЁЃ
5.2ЖдгкЪдМСКажаВЛАќКЌЃЌЕЋЖдИУЪдбщБиашЕФЪдМСзщЗжЃЌЫЕУїЪщжагІСаГіДЫРрзщЗжЕФУћГЦЁЂДПЖШЃЌЬсЙЉЯЁЪЭЛђЛьКЯЗНЗЈМАЦфЫћЯрЙиаХЯЂЁЃ
5.3ЪдМСКаШчКЌгааЃзМЦЗЛђжЪПиЦЗЃЌГ§УїШЗзщГЩГЩЗжМАЩњЮябЇРДдДЭтЃЌЛЙгІУїШЗаЃзМЦЗЫндДадЃЌЫндДадгІаДУїЫндДЕФзюИпМЖБ№ЃЌАќРЈБъзМЮяжЪБрКХЃЌжЪПиЦЗгІУїШЗАажЕЗЖЮЇЁЃ
6.ЁОДЂДцЬѕМўМАгааЇЦкЁП
6.1ЫЕУїЪдМСКаЕФаЇЦкЮШЖЈадЁЂПЊЦПЮШЖЈадЃЈШчЪЪгУЃЉЁЂИДШмЮШЖЈадЃЈШчЪЪгУЃЉЕШЃЌВЂБъУїДЂДцЬѕМўЁЃ
6.2гІзЂУїЩњВњШеЦкМАЪЙгУЦкЯоЃЌПЩМћБъЧЉЁЃ
зЂЃКДЂДцЬѕМўВЛгІгаФЃК§БэЪіЃЌШчЁАЪвЮТЁБЁЂЁАГЃЮТЁБЁЃ
7.ЁОЪЪгУвЧЦїЁП
гІУїШЗПЩЪЪгУвЧЦїЕФОпЬхЦЗХЦЁЂаЭКХЁЃ
8.ЁОбљБОвЊЧѓЁП
жиЕуУїШЗвдЯТФкШнЃКбљБОРраЭЁЂДІРэЁЂБЃДцЦкЯоМАБЃДцЬѕМўЕШЃЌШчгабЊНЌбљБОЃЌгІУїШЗПЙФ§МСЕФвЊЧѓЁЃРфВибљБОМьВтЧАЪЧЗёаыЛжИДЪвЮТЃЌРфЖГбљБОгІУїШЗЖГШкДЮЪ§ЁЃ
ЖдвбжЊИЩШХЮяНјааЫЕУїВЂУїШЗВЛВњЩњИЩШХЕФзюИпХЈЖШЃЌШчЃКЕЈКьЫиЁЂбЊКьЕААзЁЂИЪгЭШ§ѕЅЁЂПЙЛЕбЊЫсЁЃ
9.ЁОМьбщЗНЗЈЁП
ЯъЯИЫЕУїЪдбщВйзїЕФИїИіВНжшЃЌАќРЈЃК
9.1ЪдМСХфжЦЗНЗЈЁЂзЂвтЪТЯюЁЃ
9.2ЪдбщЬѕМўЃКВтЖЈЗНЗЈЃЈЗДгІРраЭЃЉЁЂЗДгІЗНЯђЁЂЮТЖШЁЂВтЖЈжї/ИБВЈГЄЁЂбљБОгУСПЁЂЪдМСгУСПЁЂПеАз/ВтЖЈЖСЪ§ЪБМфвдМАЪдбщЙ§ГЬжаЕФзЂвтЪТЯюЁЃ
9.3аЃзМЃКЫЕУїЪЙгУЕФаЃзМЦЗЃЌаЃзМЧњЯпЕФЛцжЦЗНЗЈЁЃ
9.4жЪСППижЦЃКЫЕУїжЪПиЦЗЕФЪЙгУЁЂжЪСППижЦЗНЗЈЁЂжЪПиЦЕТЪЁЃ
9.5МьбщНсЙћЕФМЦЫуЃКгІУїШЗМьбщНсЙћЕФМЦЫуЗНЗЈЁЃ
10.ЁОВЮПМЧјМфЁП
гІзЂУїбљБОРраЭЕФВЮПМЧјМфЃЌВЂМђвЊЫЕУїЦфШЗЖЈЗНЗЈЁЃНЈвщзЂУїЁАгЩгкЕиРэЁЂШЫжжЁЂадБ№КЭФъСфЕШВювьЃЌНЈвщИїЪЕбщЪвНЈСЂздМКЕФВЮПМЧјМфЁБЁЃ
11.ЁОМьбщНсЙћЕФНтЪЭЁП
ЫЕУїПЩФмЖдМьбщНсЙћВњЩњгАЯьЕФвђЫиЃЌдкКЮжжЧщПіЯТашвЊНјааШЗШЯЪдбщЃЌШчМьбщНсЙћГіЯжгыСйДВВЛЗћЩѕжСЯруЃЕФЧщПіЃЌгІЗжЮіВщевдвђВЂжиаТШЗШЯЕШЁЃ
12.ЁОМьбщЗНЗЈЕФОжЯоадЁП
ШчЪдМСКаПЩЯЁЪЭЃЌгІУїШЗЕБВтЖЈжЕГЌГіЯпадЩЯЯоЪБЕФЯЁЪЭЗНЗЈвдМАзюДѓЯЁЪЭБЖЪ§ЁЃ
ЫЕУїИУМьбщЗНЗЈЕФОжЯоадЃЌШчБОЪдМСКаЕФМьВтНсЙћНіЙЉСйДВВЮПМЃЌВЛФмзїЮЊСйДВШЗеявРОнЃЌЖдЛМепЕФСйДВеяжЮгІНсКЯЦфжЂзД/ЬхеїЁЂВЁЪЗЁЂЦфЫћЪЕбщЪвМьВщМАжЮСЦЗДгІЕШЧщПізлКЯПМТЧЕШЁЃ
13.ЁОВњЦЗадФмжИБъЁП
ЭЈГЃАќРЈвдЯТФкШнЃК
13.1ЪдМСЭтЙлЃЛ
13.2ПеАзЮќЙтЖШЃЛ
13.3ЯпадЗЖЮЇЃЛ
13.4зМШЗЖШЃЛ
13.5ЗжЮіСщУєЖШЃЛ
13.6ОЋУмЖШЃЈХњФкОЋУмЖШКЭХњМфВюЃЉЃЛ
13.7аЃзМЦЗЁЂжЪПиЦЗадФмЃЈШчЪЪгУЃЉЁЃ
14.ЁОзЂвтЪТЯюЁП
ЭЈГЃАќРЈвдЯТФкШнЃК
14.1ВЩгУВЛЭЌЗНЗЈбЇЕФЪдМСМьВтЫљЕУНсЙћВЛгІжБНгЯрЛЅБШНЯЃЌвдУтдьГЩДэЮѓЕФвНбЇНтЪЭЁЃ
14.2гаЙиШЫдДзщЗжЃЈШчгаЃЉЕФОЏИцЃЌШчЃКЪдМСКаФкЕФжЪПиЦЗЁЂаЃзМЦЗЛђЦфЫћШЫдДзщЗжЃЌЫфвбОЭЈЙ§СЫHBs-AgЁЂHIV1/2-AbЁЂHCV-AbЕШЯюФПЕФМьВтЃЌЕЋНижСФПЧАЃЌУЛгаШЮКЮвЛЯюМьВтПЩвдШЗБЃОјЖдАВШЋЃЌЙЪШдгІНЋетаЉзщЗжзїЮЊЧБдкДЋШОдДЖдД§ЁЃ
14.3ЖдЫљгабљБОКЭЗДгІЗЯЦњЮяЖМгІЪгЮЊДЋШОдДЖдД§ЁЃ
15.ЁОБъЪЖЕФНтЪЭЁП
ШчгаЭМаЮЛђЗћКХЃЌНтЪЭЦфДњБэЕФвтвхЁЃ
16.ЁОВЮПМЮФЯзЁП
гІЕБзЂУїдкБржЦЫЕУїЪщЪБЫљв§гУЕФВЮПМЮФЯзЁЃ
17.ЁОЛљБОаХЯЂЁП
17.1ОГФкЬхЭтеяЖЯЪдМС
17.1.1зЂВсШЫгыЩњВњЦѓвЕЮЊЭЌвЛЦѓвЕЕФЃЌАДвдЯТИёЪНБъзЂЛљБОаХЯЂЃК
зЂВсШЫ/ЩњВњЦѓвЕУћГЦЃЌзЁЫљЃЌСЊЯЕЗНЪНЃЌЪлКѓЗўЮёЕЅЮЛУћГЦЃЌСЊЯЕЗНЪНЃЌЩњВњЕижЗЃЌЩњВњаэПЩжЄБрКХ
17.1.2ЮЏЭаЩњВњЕФАДеевдЯТИёЪНБъзЂЛљБОаХЯЂЃК
зЂВсШЫУћГЦЃЌзЁЫљЃЌСЊЯЕЗНЪНЃЌЪлКѓЗўЮёЕЅЮЛУћГЦЃЌСЊЯЕЗНЪНЃЌЪмЭаЦѓвЕЕФУћГЦЃЌзЁЫљЃЌЩњВњЕижЗЃЌЩњВњаэПЩжЄБрКХ
17.2НјПкЬхЭтеяЖЯЪдМС
АДеевдЯТИёЪНБъзЂЛљБОаХЯЂЃК
зЂВсШЫ/ЩњВњЦѓвЕУћГЦЃЌзЁЫљЃЌЩњВњЕижЗЃЌСЊЯЕЗНЪНЃЌЪлКѓЗўЮёЕЅЮЛУћГЦЃЌСЊЯЕЗНЪНЃЌДњРэШЫЕФУћГЦЃЌзЁЫљЃЌСЊЯЕЗНЪН
18.ЁОвНСЦЦїаЕзЂВсжЄБрКХ/ВњЦЗММЪѕвЊЧѓБрКХЁП
19.ЁОЫЕУїЪщКЫзМШеЦкМАаоИФШеЦкЁП
гІзЂУїИУВњЦЗЫЕУїЪщЕФКЫзМШеЦкЁЃШчдјНјааЙ§ЫЕУїЪщЕФБфИќЩъЧыЃЌЛЙгІИУЭЌЪБзЂУїЫЕУїЪщЕФаоИФШеЦкЁЃ
Ш§ЁЂЩѓВщЙизЂЕу
ЃЈвЛЃЉММЪѕвЊЧѓжаадФмжИБъЕФЩшЖЈМАМьбщЗНЗЈЪЧЗёЗћКЯЯрЙиаавЕБъзМЕФвЊЧѓЃЛММЪѕвЊЧѓЕФИёЪНЪЧЗёЗћКЯЁЖвНСЦЦїаЕВњЦЗММЪѕвЊЧѓБраДжИЕМддђЁЗЕФЯрЙиЙцЖЈЁЃ
ЃЈЖўЃЉВњЦЗЫЕУїЪщЕФБраДФкШнМАИёЪНЪЧЗёЗћКЯЁЖЬхЭтеяЖЯЪдМСЫЕУїЪщБраДжИЕМддђЁЗЕФвЊЧѓЃЌЯрЙиФкШнЪЧЗёЗћКЯЁЖвНСЦЦїаЕЫЕУїЪщКЭБъЧЉЙмРэЙцЖЈЁЗЃЈЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжСюЕк6КХЃЉжаЖдЫЕУїЪщЕФвЊЧѓЁЃ
ЃЈШ§ЃЉЗжЮіадФмЦРЙРжИБъМАНсЙћЪЧЗёТњзуВњЦЗММЪѕвЊЧѓЕФЙцЖЈЃЛЪЧЗёТњзуБОжИЕМддђжаИїжИБъбщжЄЕФвЊЧѓЁЃ
ЃЈЫФЃЉВЮПМЧјМфШЗЖЈЪЙгУЕФЗНЗЈЪЧЗёКЯРэЃЌЪ§ОнЭГМЦЪЧЗёЗћКЯЭГМЦбЇЕФЯрЙивЊЧѓЃЌНсТлЪЧЗёКЭЫЕУїЪщЩљГЦвЛжТЁЃ
ЃЈЮхЃЉЪдМСКаЕФЮШЖЈадбаОПЗНЗЈЪЧЗёКЯРэЃЌЮШЖЈадНсТлЪЧЗёКЭЫЕУїЪщЩљГЦЕФвЛжТЁЃ
ЃЈСљЃЉСйДВЪдбщВЩгУЕФбљБОРраЭМАВЁР§ЪЧЗёТњзуЪдМСКаЩљГЦЕФдЄЦкгУЭОЃЌбљБОСПМАСйДВбаОПЕЅЮЛЕФбЁдёЁЂЖдБШЪдМСЕФбЁдёЁЂЭГМЦЗНЗЈМАбаОПНсЙћЁЂСйДВЗНАИМАБЈИцзЋаДЕФИёЪНЕШЪЧЗёЗћКЯЁЖЬхЭтеяЖЯЪдМССйДВЪдбщММЪѕжИЕМддђЁЗЖдЯрЙиФкШнЕФЙцЖЈЁЃ
ЫФЁЂБраДЕЅЮЛ
еуНЪЁвНСЦЦїаЕЩѓЦРжааФЁЃ



