ИїгаЙиЕЅЮЛЃК
БОжИЕМддђжМдкжИЕМзЂВсЩъЧыШЫЖдШЫРрУтвпШБЯнВЁЖОМьВтЪдМССйДВЪдбщЕФЩшМЦМАПЊеЙЃЌЭЌЪБвВЮЊММЪѕЩѓЦРВПУХЖдзЂВсЩъБЈзЪСЯЕФММЪѕЩѓЦРЬсЙЉВЮПМЁЃ
БОжИЕМддђЪЧеыЖдШЫРрУтвпШБЯнВЁЖОМьВтЪдМССйДВЦРМлЕФвЛАувЊЧѓЃЌЩъЧыШЫгІвРОнВњЦЗЕФОпЬхЬиадШЗЖЈЦфжаФкШнЪЧЗёЪЪгУЃЌШєВЛЪЪгУЃЌашОпЬхВћЪіРэгЩМАЯргІЕФПЦбЇвРОнЃЌВЂвРОнВњЦЗЕФОпЬхЬиадЖдзЂВсЩъБЈзЪСЯЕФФкШнНјааГфЪЕКЭЯИЛЏЁЃ
БОжИЕМддђЪЧЖдЩъЧыШЫКЭЩѓВщШЫдБЕФжИЕМадЮФМўЃЌЕЋВЛАќРЈзЂВсЩѓХњЫљЩцМАЕФааеўЪТЯюЃЌврВЛзїЮЊЗЈЙцЧПжЦжДааЃЌШчЙћгаФмЙЛТњзуЯрЙиЗЈЙцвЊЧѓЕФЦфЫћЗНЗЈЃЌвВПЩвдВЩгУЃЌЕЋашвЊЯъЯИВћУїРэгЩЃЌВЂЖдЦфПЦбЇКЯРэадНјаабщжЄЃЌЬсЙЉЯъЯИЕФбаОПзЪСЯКЭбщжЄзЪСЯЃЌЯрЙиШЫдБгІдкзёбЯрЙиЗЈЙцЕФЧАЬсЯТЪЙгУБОжИЕМддђЁЃ
БОжИЕМддђЪЧдкЯжааЗЈЙцКЭБъзМЬхЯЕвдМАЕБЧАШЯжЊЫЎЦНЯТжЦЖЈЕФЃЌЫцзХЗЈЙцКЭБъзМЕФВЛЖЯЭъЩЦЃЌвдМАПЦбЇММЪѕЕФВЛЖЯЗЂеЙЃЌБОжИЕМддђЯрЙиФкШнвВНЋЪЪЪБНјааЕїећЁЃ
вЛЁЂЪЪгУЗЖЮЇ
ШЫРрУтвпШБЯнВЁЖОЃЈHuman Immunodeficiency VirusЃЌHIVЃЉМьВтЪдМСЪЧжИРћгУУтвпбЇМАЗжзгЩњЮябЇЕШЗНЗЈдРэЖдШЫбЊЧхЁЂбЊНЌЛђЦфЫћЬхвКжаЕФЬиЖЈЕФHIVЩњЮябЇБъМЧЮяЃЌАќРЈHIV 1аЭЃЈHIV-1ЃЉp24ПЙдЁЂHIVПЙЬхЁЂHIVКЫЫсЕШНјааЖЈСПЛђЖЈадЗжЮіЕФЪдМСЁЃ
БОжИЕМддђЪЪгУгкРћгУУтвпВуЮіЗЈЁЂЛЏбЇЗЂЙтЗЈЁЂЪБМфЗжБцУтвпгЋЙтЗЈЁЂУтвпгЁМЃЗЈЕШУтвпбЇЗНЗЈЖдHIVПЙдКЭ/ЛђПЙЬхНјааЖЈадМьВтЕФЬхЭтеяЖЯЪдМСЃЌвдМАгІгУЗжзгЩњЮябЇЗНЗЈЃЈШчЪЕЪБгЋЙтОлКЯУИСДЗДгІЕШЃЉЖдHIVКЫЬЧКЫЫсНјааЖЈСПЛђЖЈадМьВтКЭЗжЮіЕФЬхЭтеяЖЯЪдМСЃЌЪЪгУгкЪзДЮзЂВсВњЦЗМАЩъЧыБфИќзЂВсЕФВњЦЗЁЃ
БОжИЕМддђВЛЪЪгУгкЙњМвЗЈЖЈбЊдДЩИВщгУHIVМьВтЪдМСЁЃ
ЖўЁЂСйДВЪдбщЩшМЦ
СйДВЪдбщЕФПЊеЙЁЂЗНАИЕФжЦЖЈвдМАБЈИцЕФзЋаДЕШОљгІЗћКЯЯрЙиЗЈЙцМАЁЖЬхЭтеяЖЯЪдМССйДВЪдбщММЪѕжИЕМддђЁЗЃЈЙњМввЉЦЗМрЖНЙмРэОжЭЈИц2021ФъЕк72КХЃЉЕФвЊЧѓЃЌШчЯрЙиЗЈЙцЁЂЮФМўгаИќаТЃЌСйДВЪдбщгІЗћКЯИќаТКѓЕФвЊЧѓЁЃ
ЃЈвЛЃЉСйДВЪдбщЩшМЦ
1.СйДВЪдбщЛњЙЙМАШЫдБ
ЩъЧыШЫгІбЁдёВЛЩйгк3МвОвНСЦЦїаЕСйДВЪдбщЛњЙЙБИАИЕФСйДВЛњЙЙПЊеЙСйДВЪдбщЁЃНЈвщдкбЁдёСйДВЕЅЮЛЪБЃЌзлКЯСїааВЁбЇБГОАЁЂHIVЕФЬиадЕШвђЫибЁдёСйДВЪдбщЛњЙЙЃЌСйДВЪдбщЛњЙЙЕФЪЕбщВйзїШЫдБгІЪьЯЄМьВтЯЕЭГЕФИїЛЗНкЃЈвЧЦїЁЂЪдМСЁЂжЪПиМАВйзїГЬађЕШЃЉЃЌЪьЯЄСйДВЪдбщЗНАИЁЃ
2.СйДВЪдбщЪЪгУШЫШККЭбљБОРраЭ
2.1ЪЪгУШЫШК
ЖдгкВЛЭЌЕФМьВтЪдМСЃЌЦфдЄЦкгУЭОВЛЭЌЃЌвђДЫШызщШЫШКврВЛЭЌЁЃЩъЧыШЫгІИљОнВЛЭЌЕФвЊЧѓШызщЪЪгУШЫШКПЊеЙСйДВЪдбщЁЃ
ЖдгкHIVПЙЬхМьВтЁЂПЙдПЙЬхСЊКЯМьВтЁЂHIVКЫЫсЖЈадМьВтЪдМСЃЌдЄЦкгУЭОЮЊHIVИаШОЕФИЈжњеяЖЯЁЃШызщШЫШКгІЮЊвЩЫЦHIVИаШОЕФШЫШКЃЌАќРЈОпгаHIVЯрЙиЕФжЂзД/ЬхеїЁЂвдМАгаЯрЙиСїааВЁбЇЪЗЕФШЫШКЁЃгІФЩШыHIVИаШОЕФВЛЭЌНјеЙНзЖЮЃЌАќРЈМБадЦкЁЂЮожЂзДИаШОЦкЁЂАЌзЬВЁЦкЁЃгІАќРЈИїИіФъСфЖЮЕФШЫШКЁЃЮЊПМВьЩъБЈВњЦЗЕФЬивьЖШЃЌСйДВЪдбщжаЛЙгІФЩШыПЩФмЛсЖдЪдМСМьВтдьГЩИЩШХЕФбљБОЃЌШчздЩэУтвпВЁЛМепЁЂРрЗчЪЊвђзгЃЈRFЃЉбєадЕФВЁР§ЁЂдаИОвдМАЦфЫћПЩФмЛсдьГЩНЛВцЗДгІЕФВЁдЬхЃЈвваЭИЮбзВЁЖОЁЂБћаЭИЮбзВЁЖОЕШЃЉИаШОЕФВЁР§ЕШЁЃЖдгкHIVПЙЬхМьВт,гІбЁдёжСЩй5ЬзОЙ§ШЋУцбщжЄЕФбєзЊбЊЧхХЬНјааМьВтЃЌЖдгкПЙдПЙЬхСЊКЯМьВтЪдМСЃЌгІбЁдёжСЩй10ЬзОЙ§ШЋУцбщжЄЕФбєзЊбЊЧхХЬНјааМьВтЃЌвдЦРМлЩъБЈВњЦЗЖдгкHIVИаШОдчЦкЕФМьГіФмСІЁЃЖдгкПЙдПЙЬхСЊКЯМьВтЪдМСЃЌЮЊГфЗжЦРМлПЙдЕФМьВтадФмЃЌгІФЩШыжСЩй10Р§ЕЅЖРПЙдбєадЕФбљБОЁЃ
Ждгк HIVКЫЫсЖЈСПМьВтЪдМСЃЌдЄЦкгУЭОЮЊЭЈЙ§ЖдЛМепбЊЧхЛђбЊНЌжаШЫРрУтвпШБЯнВЁЖОКЫЫсЛљЯпЫЎЦНКЭБфЛЏЧщПіЕФМрВтЃЌгУгкЦРЙРПЙВЁЖОжЮСЦЕФгІД№КЭжЮСЦаЇЙћЁЃШызщШЫШКгІЮЊвбШЗеяHIVИаШОЕФВЁР§ЃЌгІвде§дкНгЪмПЙВЁЖОжЮСЦЕФЛМепЮЊжїЁЃЮЊСЫШЗБЃбљБОHIVВЁЖОдиСПИВИЧЩъБЈВњЦЗЕФЯпадЗЖЮЇЃЌПЩЪЪЕБФЩШыЮДНгЪмПЙВЁЖОжЮСЦЕФЛМепЁЃШчДЫРрЪдМСдЄЦкгУЭОГ§гУгкжЮСЦМрВтЭтЃЌЛЙПЩгУгкHIVИаШОЕФИЈжњеяЖЯЃЌдђЛЙгІАДееЩЯЪіЙигкИЈжњеяЖЯдЄЦкгУЭОЕФСйДВЪдбщЩшМЦПЊеЙЯргІЕФСйДВЦРМлЁЃ
ЖдгкHIVПЙЬхШЗжЄЪдМСЃЌдЄЦкгУЭОЮЊгУгкОHIVПЙЬхЩИВщЪдбщИДМьЮЊгаЗДгІадЕФбљБОЕФШЗШЯЁЃШызщШЫШКгІвдHIVПЙЬхЩИВщЪдбщИДМьЮЊгаЗДгІадЕФВЁР§ЮЊжїЁЃЮЊСЫФмЙЛПМВьШЗжЄЪдМСЕФЬивьЖШЃЌСйДВЪдбщжагІФЩШызуЙЛЕФОШЗжЄЪдМСМьВтЮЊвѕадЕФШЫШКЁЃ
ЮвЙњОГФкСїааЕФHIVжївЊЮЊHIV-1аЭMзщЃЌЦфГЃМћЕФЛљвђаЭжївЊЮЊB/BЁЏЁЂBCжизщаЭЃЈАќРЈCRF 07_BCжизщаЭКЭCRF 08_BCжизщаЭЃЉвдМАAEжизщаЭЃЈCRF 01_AEжизщаЭЃЉЁЃдкбЁдёHIVИаШОепВЁР§ЪБЃЌЪзЯШгІИљОнHIVСїааЕФЧщПіЃЌбЁдёФмДњБэЮвЙњВЛЭЌЕиЧјСїааЛљвђаЭЕФHIV-1ИаШОепВЁР§ЃЌвдЖдЪдМСМьВтЮвЙњСїааЕФHIV-1ВЁЖОЕФФмСІНјааПЭЙлПЦбЇЕФЦРМлЃЌбЁдёЕФЛљвђаЭгІжСЩйАќРЈЩЯЪіШ§жжжївЊЕФЛљвђаЭЃЌЧвУПжжЛљвђаЭгІжСЩй30Р§ЁЃЖдгкЮвЙњОГФкКБМћЕФЛљвђаЭЃЈ1аЭOзщЁЂ2аЭЃЉЃЌгІОЁСПФЩШыЃЌШчСйДВЪдбщжаЮДЖд1аЭOзщЁЂ2аЭНјаабщжЄЃЌЩъЧыШЫдкСйДВЧАадФмбаОПжаНјааСЫГфЗжбаОПЕФЧАЬсЯТЃЌПЩдкВњЦЗЫЕУїЪщЕФМьбщЗНЗЈЕФОжЯоаджаНјааЫЕУїЁЃ
2.2бљБОРраЭ
бљБОРраЭвЛАуЮЊбЊЧхЁЂбЊНЌЁЃЖдгкПкЧЛеГФЄЩјГівКЁЂФђвКЕШЬхвКбљБОЃЌгІЕЅЖРНјааСйДВЦРМлЁЃ
СйДВбљБОЕФДІРэЁЂБЃДцКЭКЫЫсЬсШЁЕШгІЗжБ№ТњзуЩъБЈВњЦЗЫЕУїЪщМАЖдБШЪдМСЫЕУїЪщЕФЯрЙивЊЧѓЁЃ
3.ЖдБШЗНЗЈЕФбЁдё
ЖдгквбгавбЩЯЪаЭЌРрВњЦЗЕФЪдМСЃЌПЩбЁдёОГФквбХњзМЩЯЪаЁЂСйДВЦеБщШЯЮЊжЪСПНЯКУЕФЭЌРрВњЦЗзїЮЊЖдБШЪдМСЃЌЪЙЩъБЈВњЦЗгыжЎНјааЖдБШЪдбщбаОПЃЌПМВьЩъБЈВњЦЗгыЖдБШЪдМСЕФвЛжТадЁЃЖдБШЪдМСЕФбЁдёгІДгдЄЦкгУЭОЁЂбљБОвЊЧѓЁЂМьВтадФмЕШЗНУцЃЌШЗШЯЦфгыЩъБЈВњЦЗОпгаНЯКУЕФПЩБШадЁЃ
ЖдгкЮовбЩЯЪаЭЌРрВњЦЗЕФаТHIVМьВтЪдМСЃЌЦфЖдБШЗНЗЈгІбЁдёгыСйДВВЮПМБъзМНјааБШНЯбаОПЁЃСйДВВЮПМБъзММДВЮееСйДВЙЋШЯЕФСйДВеяСЦжИФЯЁЂАЌзЬВЁеяЖЯБъзМЕШНјааВЁР§еяЖЯЕФЗНЗЈЁЃЖдгкЖЈСПМьВтЪдМСЭЌЪБЛЙгІЖдЦфСПжЕгыСйДВНјеЙзДПіЁЂзЊЙщЁЂжЮСЦЧщПіЕШЕФЯрЙиадНјааЗжЮіЁЃ
4.зюЕЭбљБОСП
СйДВЪдбщбљБОСПгІВЩгУЪЪЕБЕФЭГМЦбЇЗНЗЈНјааЙРЫуЃЌВЂЯъЯИУшЪіЫљЪЙгУЭГМЦЗНЗЈМАИїВЮЪ§ЕФШЗЖЈвРОнЁЃ
ЖдгкПЙЬхМьВтЁЂПЙдПЙЬхСЊКЯМьВтвдМАКЫЫсМьВтЖЈадМьВтЪдМСЃЌДЫВПЗжСйДВЪдбщФПЕФЮЊЦРЙРСНжжМьВтЗНЗЈжЎМфЕФвЛжТадЃЌвђДЫНЈвщВЩгУЕЅзщФПБъжЕЗЈНјаазюЕЭбљБОСПЕФЙРЫуЁЃЭЈЙ§бєадЗћКЯТЪКЭвѕадЗћКЯТЪРДЗжБ№МЦЫуЫљашбєадбљБОКЭвѕадбљБОЕФР§Ъ§ЁЃвѕЁЂбєадЗћКЯТЪЕФСйДВПЩНгЪмБъзМЃЈP0ЃЉНЈвщВЛЕЭгк98%ЁЃ
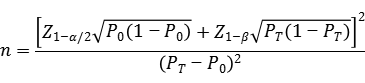
ЙЋЪНжаЃЌnЮЊбљБОСПЃЛZ1-ІС/2ЁЂZ1-ІТЮЊЯджјадЫЎЦНКЭАбЮеЖШЕФБъзМе§ЬЌЗжВМЕФЗжЪ§ЮЛЃЌP0ЮЊЦРМлжИБъЕФСйДВПЩНгЪмБъзМЃЌPTЮЊЩъБЈВњЦЗЦРМлжИБъдЄЦкжЕЁЃ
ЖдгкКЫЫсЖЈСПМьВтЪдМСЃЌвВгІбЁдёКЯЪЪЕФбљБОСПЙРЫуЕФЗНЗЈНјаазюЕЭбљБОСПЕФЙРЫуЁЃгІзЂвтЫљФЩШыЕФСйДВбљБОгІИВИЧЩъБЈВњЦЗЕФЯпадЗЖЮЇЁЃ
5. ЭГМЦЗжЮі
5.1ЖЈадМьВтЪдМС
гІбЁдёКЯЪЪЕФЭГМЦЗНЗЈЖдСйДВЪдбщНсЙћНјааЭГМЦЗжЮіЃЌЖдгкЩъБЈВњЦЗгыЖдБШЗНЗЈЕФвЛжТадЦРМлЃЌГЃбЁдё2ЁС2БэЕФаЮЪНзмНсСНжжЗНЗЈЕФНсЙћЃЌЦРМлжИБъвЛАуАќРЈбєадЗћКЯТЪ/СщУєЖШЁЂвѕадЗћКЯТЪ/ЬивьЖШЃЌKappaжЕЕШЃЌВЂМЦЫуЯргІЕФ95%жУаХЧјМфЁЃжУаХЧјМфЕФЯТЯогІТњзуЫљЩшЖЈЕФP0ЕФвЊЧѓЁЃ
ЖдгкСНжжЗНЗЈМьВтВЛвЛжТЕФбљБОЃЌгІВЩгУСйДВВЮПМБъзМЛђЦфЫћЗНЗЈдйДЮНјааШЗШЯЛђЬсЙЉСйДВеяЖЯзЪСЯвдНјвЛВНУїШЗбљБОЃЈВЩМЏбљБОЪБЪмЪдепЃЉЫљДІЕФИаШОзДЬЌЃЌДгЖјЖдЩъБЈВњЦЗЕФадФмНјааПЭЙлПЦбЇЕФЦРМлЁЃ
5.2КЫЫсЖЈСПМьВтЪдМС
5.2.1гІИљОнСйДВЪдбщЪ§ОнЛцжЦЩЂЕуЭМЃЌНјааЯрЙиадЗжЮіЁЃ
5.2.2ВЩгУBland-AltmanЗЈЃЌМЦЫувЛжТадЯоЖШЃЌЦРМлСНжжМьВтНсЙћЕФвЛжТадЁЃвЛжТадЯоЖШгІдкСйДВЫљФмНгЪмЕФНчжЕЗЖЮЇФкЁЃ
5.2.3 ВЩгУЛиЙщЗжЮіЖдСНжжМьВтЗНЗЈЕФвЛжТадНјааЦРМлЁЃгІИљОнЪ§ОнЗжВМЬиЕуЕШвђЫибЁдёЪЪгУЕФЛиЙщЗжЮіЗНЗЈЃЌШчPassing-BablokЛиЙщЁЂDemingЛиЙщКЭзюаЁЖўГЫЛиЙщЕШЁЃЛиЙщЗжЮігІжиЕуЙлВьЛиЙщЗНГЬЕФЛиЙщЯЕЪ§КЭНиОрЕШжИБъЃЌМЦЫуЛиЙщЯЕЪ§КЭНиОрЕФ95%жУаХЧјМфЁЃ
ЃЈЖўЃЉЭЈгУвЊЧѓ
1.СйДВЪдбщЗНАИ
СйДВЪдбщЪЕЪЉЧАЃЌбаОПШЫдБгІДгСїааВЁбЇЁЂЭГМЦбЇЁЂСйДВвНбЇЁЂМьбщвНбЇЕШЖрЗНУцПМТЧЃЌЩшМЦПЦбЇКЯРэЕФСйДВЪдбщЗНАИЁЃИїСйДВЪдбщЛњЙЙгІжДааЭГвЛЕФСйДВЪдбщЗНАИЃЌЧвБЃжЄдкећИіСйДВЪдбщЙ§ГЬжазёбдЄЖЈЕФЗНАИЪЕЪЉЃЌВЛПЩЫцвтИФЖЏЁЃећИіЪдбщЙ§ГЬгІдкСйДВЪдбщЛњЙЙЕФЪЕбщЪвФкВЂгЩБОЪЕбщЪвЕФММЪѕШЫдБВйзїЭъГЩЃЌЩъЧыШЫЕФММЪѕШЫдБГ§НјааБивЊЕФММЪѕжИЕМЭтЃЌВЛЕУЫцвтИЩЩцЪЕбщНјГЬЃЌгШЦфЪЧЪ§ОнЪеМЏЙ§ГЬЁЃ
ЪдбщЗНАИжагІШЗЖЈбЯИёЕФВЁР§ФЩШы/ХХГ§БъзМЃЌШЮКЮвбОШыбЁЕФВЁР§дйБЛХХçóСйДВЪдбщЖМгІМЧТМдкАИВЂУїШЗЫЕУїдвђЁЃдкЪдбщВйзїЙ§ГЬжаКЭХаЖЈЪдбщНсЙћЪБгІВЩгУУЄЗЈвдБЃжЄЪдбщНсЙћЕФПЭЙладЁЃ
гІзЂвтЃЌЖдгкHIVПЙЬхМьВтЪдМСЁЂПЙдПЙЬхМьВтЪдМСЃЌЖдгкГѕМьгаЗДгІадЕФбљБОгІНјааИДМьЃЌвђДЫСйДВЪдбщжагІбЯИёАДееЫЕУїЪщЕФвЊЧѓЖдГѕМьгаЗДгІадЕФбљБОНјааИДМьЃЌЪдбщНсЪјКѓгІАДееИДМьЕФНсЙћФЩШыЭГМЦЗжЮіЁЃЫЕУїЪщжагІгаЙигкИДМьЕФвЊЧѓЁЃ
2.жЪСППижЦ
СйДВЪдбщПЊЪМЧАЃЌНЈвщНјааСйДВЪдбщЕФдЄЪдбщЃЌвдЪьЯЄВЂеЦЮеЯрЙиЪдбщЗНЗЈЕФВйзїЁЂвЧЦїЁЂММЪѕадФмЕШЃЌзюДѓЯоЖШПижЦЪдбщЮѓВюЁЃећИіЪдбщЙ§ГЬЖМгІДІгкгааЇЕФжЪСППижЦЯТЃЌзюДѓЯоЖШБЃжЄЪдбщЪ§ОнЕФзМШЗадМАОЋУмЖШЁЃ
3.СйДВЪдбщБЈИц
СйДВЪдбщБЈИцгІИУЖдЪдбщЕФећЬхЩшМЦМАИїИіЙиМќЕуИјгшЧхЮњЁЂЭъећЕФВћЪіЃЌгІИУЖдећИіСйДВЪдбщЪЕЪЉЙ§ГЬЁЂНсЙћЗжЮіЁЂНсТлЕШНјааЬѕРэЗжУїЕФУшЪіЃЌВЂгІАќРЈБивЊЕФЛљДЁЪ§ОнКЭЭГМЦЗжЮіЗНЗЈЃЌзюКѓЕУГіСйДВЪдбщНсТлЁЃСйДВЪдбщБЈИцЕФзЋаДВЮПМЁЖЬхЭтеяЖЯЪдМССйДВЪдбщММЪѕжИЕМддђЁЗМАЁЖЬхЭтеяЖЯЪдМССйДВЪдбщБЈИцЗЖБОЁЗЕФЯрЙивЊЧѓЁЃ
Ш§ЁЂВЮПМЮФЯз
[1]ЬхЭтеяЖЯЪдМСзЂВсгыБИАИЙмРэАьЗЈЃЈЙњМвЪаГЁМрЖНЙмРэзмОжСюЕк48КХЃЉ[Z].
[2]ЙигкЙЋВМЬхЭтеяЖЯЪдМСзЂВсЩъБЈзЪСЯвЊЧѓКЭХњзМжЄУїЮФМўИёЪНЕФЙЋИцЃЈЙњМввЉЦЗМрЖНЙмРэОжЙЋИц2021ФъЕк122КХЃЉ[Z].
[3]ЬхЭтеяЖЯЪдМССйДВЪдбщММЪѕжИЕМддђЃЈЙњМввЉЦЗМрЖНЙмРэОж2021ФъЕк72КХЃЉ[Z].
[4]ЬхЭтеяЖЯЪдМССйДВЪдбщБЈИцЗЖБОЃЈЙњМввЉЦЗМрЖНЙмРэОж2022ФъЕк21КХЃЉ[Z].
[5]ЬхЭтеяЖЯЪдМСЫЕУїЪщБраДжИЕМддђЃЈдЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙЋИц2014ФъЕк17КХЃЉ[Z].
[6]WS-293-2019,АЌзЬВЁКЭАЌзЬВЁВЁЖОИаШОеяЖЯБъзМ [S].
[7]жаЛЊвНбЇЛсИаШОВЁбЇЗжЛсАЌзЬВЁБћаЭИЮбзбЇзщ жаЙњМВВЁдЄЗРПижЦжааФ.жаЙњАЌзЬВЁеяСЦжИФЯЃЈ2021АцЃЉ[J],жаЙњАЌзЬВЁадВЁ, 2021,27(11):1182-1201.
[8]жаЙњМВВЁдЄЗРПижЦжааФ.ШЋЙњАЌзЬВЁМьВтММЪѕЙцЗЖЃЈ2020ФъаоЖЉАцЃЉ[Z].



